Creutzfeldt-Jakob Disease
by Yasmin Mamani, MBS 2020, GCSoM
Mentor: Brian Piper, PhD
A patient in his fifties arrives at emergency department unable to walk in a straight line, slurring his speech, and unaware of his name or spatial orientation. Upon first glance, one might think he’s intoxicated! The patient is confused and obviously agitated; even his daughter cannot calm him down. However, a urine analysis revealed no alcohol or drugs in his system. His daughter recounts no history of substance misuse, and that his sudden decline in function occurred only in the past week. Could he have suffered a stroke? As more of his family begins to arrive, his nurses and physician learn that their patient has been forgetful and confused for three-months. Ah, ha! His age, all of his symptoms, and patient history points to dementia, one of the most commonly diagnosed neurological disorders.
Unfortunately, nobody in this situation would have thought that this man suffered from one of the rarest neurodegenerative diseases known to medicine, one that afflicts approximately 1 in a million. Sadly, the patient passes away at the hospital within three weeks of arriving. An autopsy reveals that he died from Creutzfeldt-Jakob Disease (CJD).
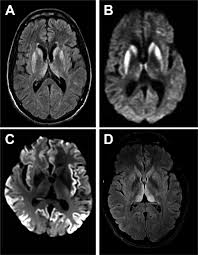
CJD is a neurological disease that causes the brain to assume a spongy, porous appearance. Spongy brains are the hallmark feature for a deadly group of disorders known as prion diseases, to which CJD belongs. As important regions of the brain are lost to these developing holes, patients experience a sharp decline in their thinking, motor skills, and overall health. Many of the symptoms of CJD resemble Alzheimer’s disease, Parkinson’s disease, Amyotrophic lateral sclerosis, and dementia disorders. Unfortunately, CJD is always fatal and there is no cure. Even more terrifying, there is no verified examination that can positively diagnose someone with CJD.
What causes CJD? Proteins in the central nervous system misfold into a configuration that cannot be digested. As the undigestible proteins accumulate, they begin to form tangles that choke out surrounding neurons. Neurons can be thought of as the main component of the nervous system; they are the worker ants that keep our brains functioning. These protein tangles cause neurons to die off in chunks, forming holes in the brain. Dead neurons cannot communicate with each other, so patients quickly begin to experience problems thinking, remembering, performing daily tasks, and controlling their emotions. Eventually, all CJD patients require palliative care.
Axons can be thought of the electrical wires that connect neurons; they transmit signals about the outside environment to the brain for interpretation. When axons break down, they release small fragments into the blood and fluid of the brain. Researchers run samples of blood or brain fluid to look for these fragments, also known as neurofilament light chains, because their presence in high numbers indicates that many axons have broken down. Currently, scientists have been looking for a type of biological marker that can point to the presence of prions. Neurofilament light chains have shown promising results, although there is still much research to be conducted. Studies show that neurofilament light chains may be an excellent tool to distinguish between the different subtypes of CJD, such as genetic CJD, sporadic CJD, and iatrogenic CJD. Surveillance of CJD relies upon understanding the etiologic origin of a case, which can have significant implications for families and patients involved. For example, what does it mean for the children or grandchildren of a patient diagnosed with genetic CJD? How do family members move forward with the possibility that they might carry the mutation causing CJD? Without biomarkers like neurofilament light chains, clinicians might never be able to answer such questions, nor could they provide the most precise treatment plan for their patient.
Instead of testing something new, some researchers have also considered refining laboratory tests that have worked for years. Real-time quaking induced conversion assays are commonplace in clinical settings, and some investigations have used them to detect prions. RT-QuIC assays can take a suspicious sample of brain fluid and expose it to a solution containing normal, properly folded protein substrates. Heating and mixing the combination on a laboratory machine will speed up the pace at which prions will attack the normal proteins, telling scientists how many prions are present in the sample.
Our patient, misdiagnosed and rapidly fading, could only benefit from palliative care. Since there is no screening test and no cure, patients and their families are often left frustrated and upset, desperate for an answer. Research is moving forward, exploring the applications of neurofilament light chains and RT-QuIC assays, but it will be many years until the answer is found. A reliable, accurate, and inexpensive test is urgently needed for patients suffering from CJD.


Comments
Post a Comment